Cooperative Model Framework with Minimal Viable Theoretical Data
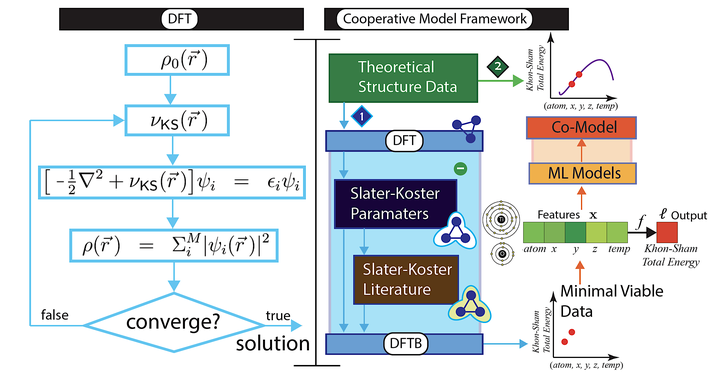
Predicting material properties by solving the Kohn‐Sham (KS) equation, which is the basis of modern computational approaches to electronic structures, has provided significant improvements in materials sciences. Despite its contributions, both DFT and DFTB calculations are limited by the number of electrons and atoms that translate into increasingly longer run‐times. In this work we introduce a novel, data‐centric machine learning framework that is used to rapidly and accurately predicate the KS total energy of anatase TiO2 nanoparticles (NPs) at different temperatures using only a small amount of theoretical data. The proposed framework that we call co‐modeling eliminates the need for experimental data and is general enough to be used over any NPs to determine electronic structure and, consequently, more efficiently study physical and chemical properties. We include a web service to demonstrate the effectiveness of our approach.